C’est l’histoire d’une femme de 31 ans, non obèse (IMC : 21 kg/m2) à laquelle on vient de diagnostiquer à plusieurs reprises une légère hyperglycémie dans le cadre d’un dépistage de la médecine du travail. Son hémoglobine glyquée (HbA1c), témoin du niveau moyen de la glycémie des 4 à 6 semaines précédant le prélèvement, est normale (5,7 %).
Elle n’a pas souvenir d’avoir ressenti de symptômes d’hyperglycémie, ou d’hypoglycémie, pendant son enfance. Il n’y a pas d’antécédents familiaux de diabète, mais deux membres de la fratrie sont obèses.
Diabète gestationnel insulino-dépendant
Cette patiente va présenter un diabète gestationnel, défini par un trouble de la tolérance glucidique conduisant à une hyperglycémie diagnostiquée au cours de la grossesse. En effet, deux ans plus tard, lors d’une première grossesse, les analyses de sang et une épreuve d’hyperglycémie provoquée montrent un taux élevé de glucose sanguin. Un traitement par sulfamide hypoglycémiant (glibenclamide) est commencé, qui l’aide à contrôler sa glycémie. Mais l’état de la patiente nécessite par la suite un traitement par insuline.
La glycémie s’améliore après cette première grossesse, mais ne revient pas à la normale. Lors des grossesses suivantes, la patiente a toujours besoin d’avoir recours à l’insuline. L’insulinothérapie est interrompue six mois après la naissance du troisième enfant, mais la patiente continue de présenter une hyperglycémie car sa glycémie à jeun est à 7,3 mmol/L, (considérée normale si elle est comprise entre 4 et 5,83 mmol/L) et son HbA1c est à 6,5 %, compatible avec un diagnostic de diabète. Elle est alors traitée par deux antidiabétiques oraux, le glibenclamide et la metformine. L’hémoglobine glyquée (HbA1c) descend à 6,1 %.
Le temps passé dans la cible glycémique, c’est-à-dire le pourcentage de temps passé dans la plage de valeurs normales de la glycémie, est de plus de 88 %. La glycémie s’en écarte environ 2 % du temps en étant trop basse (hypoglycémie) et 9 % du temps en étant trop haute (hyperglycémie). L’année suivante, l’hémoglobine glyquée est à 7,3 % alors que la patiente, alors âgée de 38 ans, est toujours sous glibenclamide et metformine.
Les trois enfants sont nés par césarienne et pesaient respectivement 3,4 kg, 4,4 kg et 3,9 kg à la naissance.
Compte tenu du jeune âge de la patiente (31 ans) au moment du diagnostic d’hyperglycémie, de l’absence d’anticorps anti-îlots, de l’absence d’obésité, les médecins pensent que cette femme présente un diabète insulino-dépendant d’origine génétique. À l’âge de 35 ans, la patiente se soumet à un examen génétique destiné à rechercher la présence de mutations dans 14 gènes différents associés à un diabète MODY (maturity onset diabetes of the young), diabète de la maturité apparaissant chez le jeune.
Analyse génétique
Le séquençage génétique révèle la présence d’une mutation dans le gène KCNJ11, situé sur le chromosome 11.
Il s’agit d’une mutation hétérozygote : une seule des deux copies de ce gène est mutée. L’existence de ce variant génétique (p.Ser118Leu, caractérisé par le remplacement de l’acide aminé sérine par une leucine) n’a été décrit qu’une seule fois auparavant, sans que l’on connaisse son impact sur la fonction de la protéine mutée.
Mutation sur un gène codant le canal potassique ATP-dépendant de la cellule pancréatique sécrétrice d’insuline
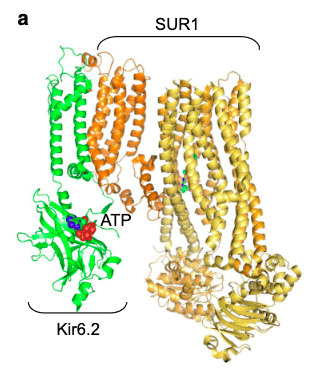
La membrane des cellules bêta du pancréas possède des canaux sélectifs pour le potassium directement inhibés par l’ATP intracellulaire : ce sont les canaux potassiques ATP-dépendants. Les canaux KATP, constitués des sous-unités Kir6.2 et SUR1, sont considérés comme la cible principale du glucose pour le contrôle de la sécrétion d’insuline. Leur fermeture provoque une dépolarisation, puis une augmentation de la concentration intracellulaire de calcium qui déclenche la sécrétion d’insuline. Le bon fonctionnement du canal KATP nécessite une communication sans faille entre ses deux composants Kir6.2 et SUR1.
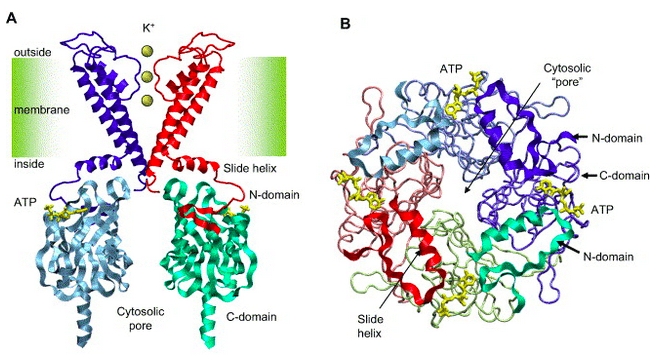
Pour comprendre la suite, il convient de dire un mot du canal potassique de la cellule bêta pancréatique, sécrétrice d’insuline. En effet, dans cette cellule, le canal potassique, sensible à l’ATP (molécule énergétique), joue un rôle crucial dans la stimulation de la sécrétion d’insuline en réponse au glucose. On parle de canal potassique car celui-ci forme un pore qui traverse la membrane plasmique de la cellule et laisse passer les ions potassium (K+).
Ce pore est composé de quatre sous-unités internes (Kir6.2), dont chacune possède un site de fixation pour l’ATP. Ce sont ces sous-unités qui permettent le passage des ions potassium, assurant donc la conductance potassique proprement dite. Le canal potassique comporte également quatre autres sous-unités externes, dites régulatrices (SUR1).
La sous-unité Kir6.2 du canal potassique est codée par le gène KCNJ11, celui-là même qui est muté chez notre patiente trentenaire, diabétique et mère de trois enfants. La synthèse de la sous-unité régulatrice SUR1 dépend, elle, du gène ABCC8.
Les mutations touchant la sous-unité Kir6.2 du canal potassique peuvent être divisées en deux catégories. D’une part, les mutations gain de fonction, qui diminuent la sécrétion d’insuline et causent une hyperglycémie. Dans ce cas, les canaux sont suractivés. D’autre part, les mutations perte de fonction, qui ont pour conséquence l’augmentation de la sécrétion d’insuline et induisent une hypoglycémie.
Les mutations du gène KCNJ11 ont pour conséquence de maintenir de manière inappropriée les canaux potassiques ouverts et ce, même en cas d’hyperglycémie. En l’absence de fermeture des canaux, la membrane cellulaire n’est pas en mesure de se dépolariser efficacement, ce qui l’empêche d’émettre un signal déclenchant la libération d’insuline par la cellule bêta pancréatique.
Les mutations du gène KCNJ11 entraînent habituellement la survenue d’un diabète néonatal, avant l’âge de six mois, parfois plus tard.
Les chercheurs ont entrepris de déterminer si cette mutation faux-sens identifiée dans l’une des deux copies du gène KCNJ11 pouvait expliquer le diabète de la patiente, sa réponse aux sulfamides hypoglycémiants et son manque de résistance à l’insuline.
Analyses fonctionnelles
Pour ce faire, ils ont analysé l’impact de la mutation S118L sur la fonction du canal potassique. Malheureusement, n’ayant pu avoir accès à leur génome, ils n’ont pas pu déterminer si cette mutation était également présente chez les trois enfants de la patiente. Ils savaient néanmoins que la glycémie de chaque enfant se situait dans les valeurs normales.
Les résultats des expériences réalisées par des biologistes de l’université d’Oxford, de Chicago et de Memphis sont publiés dans un article paru le 17 février 2024 dans la revue Diabetologia. Les chercheurs ont utilisé des cellules rénales et des ovocytes de xénope (modèle amphibien de laboratoire) dans lesquels ils ont introduit un ADN porteur du gène KCNJ11 normal ou porteur de la mutation S118L. Les biologistes cellulaires ont ensuite étudié l’excitabilité membranaire de ces mêmes cellules, autrement dit la production de courants électriques à leur surface. Ils ont enfin évalué l’expression du gène muté à la surface des cellules, à savoir le nombre de canaux potassiques présents sur leur membrane extérieure.
Ces analyses fonctionnelles ont révélé que la sensibilité du canal potassique muté à l’ATP est inchangée. En revanche, ils ont observé une réduction de 40 % de la présence du canal potassique muté à la surface des cellules, en comparaison avec celles porteuses de ce même canal non muté. Il ne s’agit donc pas d’une mutation gain de fonction, mais d’une mutation perte de fonction qui réduit l’expression membranaire du canal potassique.
Le site de la mutation S118L se situe dans la région du pore du canal potassique, tout près de la sous-unité Kir6.2, mais à bonne distance de la sous-unité SUR1. Il est donc possible que cette mutation affecte l’assemblage des composants du canal en désorganisant les interactions entre les sous-unités Kir6.2, et indirectement celles entre les sous-unités Kir.6.2 et SUR1.
Ce résultat indique que la mutation S118L perturbe le trafic et/ou l’assemblage des sous-unités composant le canal potassique. Tout se passe donc comme si la mutation empêchait l’exportation de ce canal vers la surface des cellules, perturbant ce que l’on appelle le trafic membranaire.
Le glibenclamide, un sulfamide hypoglycémiant, corrige l’effet de la mutation
Or il se trouve que celui-ci est restauré lorsque les cellules sont cultivées en présence de glibenclamide, un sulfamide hypoglycémiant. En d’autres termes, cet antidiabétique oral corrige l’effet de la mutation S118L du gène KCNJ11 en augmentant l’expression des canaux potassiques mutés à la surface cellulaire. Il est possible que le glibenclamide agisse en augmentant le temps durant lequel les sous-unités Kir6.2 et SUR1 interagissent entre elles, ce qui favoriserait leur assemblage et permettrait de corriger le trafic membranaire.
En général, les mutations perte de fonction provoquent des réductions comparables de la densité des canaux potassiques et s’accompagnent d’un hyperinsulinisme congénital, pas d’un diabète, soulignent les auteurs de l’étude dirigée par Frances Ashcroft (université d’Oxford). Par ailleurs, la pathologie associée à ce type de mutation s’exprime de manière variable selon les individus. On parle de maladie de transmission autosomique dominante avec une pénétrance faible. Ainsi, certains individus porteurs d’une mutation perte de fonction sur ce canal potassique peuvent ne pas présenter de symptômes d’hyperinsulinisme, ou alors ceux-ci peuvent passer inaperçus. De plus, on observe une disparition spontanée des symptômes, qui survient dans de nombreux cas. Ceci pourrait expliquer qu’un diagnostic d’hyperinsulinisme n’ait pas été posé chez cette patiente, font remarquer les auteurs de l’étude.
Ces chercheurs évoquent cependant une autre hypothèse : la mutation S118L du gène KCNJ11 pourrait ne pas provoquer de symptômes d’hyperinsulinisme (hypoglycémie) durant l’enfance, mais prédisposer à une intolérance au glucose et au diabète au début de l’âge adulte. Cette possibilité semble être celle qui correspond le mieux aux données de la patiente. De plus, d’autres éléments plaident pour cette hypothèse. L’histoire clinique de la patiente ressemble à celle de certaines personnes porteuses d’une mutation (E1506K) du gène ABCC8, celui qui code pour la sous-unité régulatrice SUR1 du canal potassique de la cellule bêta pancréatique. Certaines de ces patientes présentent en effet un diabète gestationnel qui disparaît après la grossesse et réapparaît ultérieurement.
Par ailleurs, des patients, hommes et femmes, porteurs d’une autre mutation perte de fonction (R1353H) du gène ABCC8, déjà connue pour causer un hyperinsulinisme congénital, ont été diagnostiqués avec un hyperinsulinisme, un diabète gestationnel et un diabète avec absence d’auto-anticorps.
Enfin, ceci n’est pas sans rappeler ce que l’on observe chez des patients atteints de diabète MODY3, caractérisé par une mutation du gène HNF1A (hepatocyte nuclear factor 1 alpha) et chez ceux ayant un diabète MODY1, dû à des mutations du gène HNF4A (hepatocyte nuclear factor 4 alpha). HNF1A et HNF4A sont des facteurs de transcription qui régulent l’expression de nombreux gènes. Chez ces patients atteints de MODY-HNF1A et MODY-HNF4A, on observe un hyperinsulinisme à la naissance, puis une diminution de la sécrétion d’insuline et un diabète plus tard dans la vie, le plus souvent au moment de la puberté.
Que retenir de ce cas clinique ? Tout d’abord que la cause du diabète de cette patiente, diagnostiqué au début de l’âge adulte, n’a pu être identifiée que par l’analyse génétique, les études fonctionnelles révélant que l’anomalie identifiée est une mutation perte de fonction. Ensuite que la découverte de cette mutation a permis à cette jeune patiente d’être traitée par un sulfamide hypoglycémiant par voie orale (associé au semaglutide) plutôt que par l’insuline, ce qui est bien moins contraignant.
En 2008, une équipe israélienne avait également pu traiter une petite fille par glibenclamide après avoir identifié une mutation perte de fonction dans la sous-unité SUR1. Elle avait présenté un hyperinsulinisme dans l’enfance avant de développer un diabète manifeste à l’âge de dix ans et demi.
Ce cas clinique illustre donc l’importance de l’analyse génétique dans le diagnostic d’un diabète atypique. Les individus présentant un diabète de survenue précoce devraient être testés pour des mutations dans les gènes KCNJ11 et ABCC8 car ils ont pu présenter antérieurement un hyperinsulinisme non détecté. Porter un diagnostic précis est important pour le patient et sa famille, car cela permet d’établir une prise en charge thérapeutique optimale, adaptée à l’âge du patient et au gène impliqué, autrement dit de pratiquer une médecine de précision.
Marc Gozlan (Suivez-moi sur X, Facebook, LinkedIn, Mastodon, Bluesky)